Title
Phase III study of Isatuximab-Carfilzomib-Lenalidomide-Dexamethasone (Isa-KRd) versus Carfilzomib-Lenalidomide-Dexamethasone (KRd) in newly diagnosed multiple myeloma patients eligible for autologous stem cell transplantation (IsKia trial)Study map
Overview / Summary
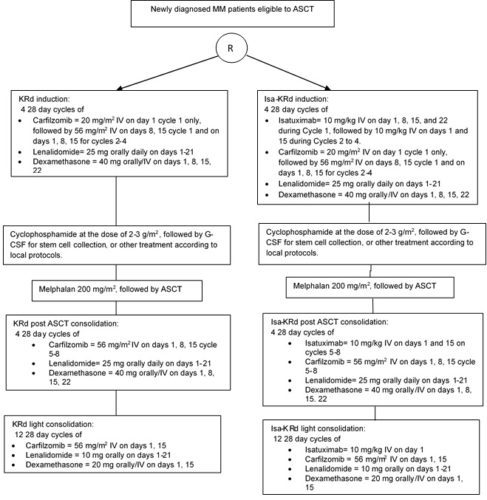
This protocol is a phase III study designed to compare the efficacy and the safety of Isa-KRd induction, transplant, Isa-KRd post ASCT consolidation and Isa-KRd light consolidation vs KRd induction, transplant, KRd post ASCT consolidation and KRd light consolidation
After confirmation of eligibility criteria patients will be randomized to one of the 2 treatment groups in a 1:1 randomization ratio: Isatuximab-carfilzomib-lenalidomide-dexamethasone (ARM A) or carfilzomib-lenalidomide-dexamethasone (ARM B) for 4 cycles followed by ASCT. Each arm will receive then 4 cycles of post ASCT consolidation followed by 12 cycles of light consolidation with isatuximab-carfilzomib-lenalidomide-dexamethasone (ARM A) or carfilzomib-lenalidomide-dexamethasone (ARM B)
Study details
Patient eligibility criteria
Inclusion criteria
- Patient with newly diagnosed multiple myeloma and eligible to ASCT.
- Patient is, in the investigator’s opinion, willing and able to comply with the study visits and procedures required per protocol.
- Patient has provided written informed consent in accordance with federal, local, and institutional guidelines prior to initiation of any study-specific activities or procedures. Subject does not have kind of condition that, in the opinion of the Investigator, may compromise the ability of the subject to give written informed consent and patient is, in the investigator(s) opinion, willing and able to comply with the protocol requirements.
- Monoclonal plasma cells in the bone marrow ≥10% or presence of a biopsy proven plasmacytoma and documented multiple myeloma satisfying at least one of the calcium, renal, anemia, bone (CRAB) criteria or biomarkers of malignancy criteria:
CRAB criteria:
– Hypercalcemia: serum calcium >0.25 mmol/L (>1 mg/dL) higher than upper limit of normal (ULN) or >2.75 mmol/L (>11 mg/dL)
– Renal insufficiency: creatinine clearance <40mL/min or serum creatinine >177 μmol/L (>2 mg/dL)
– Anemia: hemoglobin >2 g/dL below the lower limit of normal or hemoglobin <10 g/dL
– Bone lesions: one or more osteolytic lesions on skeletal radiography, CT, or PET-CT
Biomarkers of Malignancy:
– Clonal bone marrow plasma cell percentage ≥60%
– Involved: uninvolved serum FLC ratio ≥100
– >1 focal lesion on magnetic resonance imaging (MRI) studies - Patient is 18 – 70 years old and is eligible for autologous stem cell transplantation
- Patient has measurable disease as defined by any one of the following:
– Serum monoclonal paraprotein (M-protein) level ≥1.0 g/dL or urine M-protein level ≥200 mg/24 hours; or
– Light chain multiple myeloma without measurable disease in the serum or the urine: Serum immunoglobulin FLC ≥10 mg/dL and abnormal serum immunoglobulin kappa lambda FLC ratio. - Life expectancy ≥ 3 months
- ECOG status ≤2
- Clinical laboratory values meeting the following criteria during the Screening Phase:
- Adequate hepatic function, with serum (alanine aminotransferase) ALT ≤ 2.5 times the upper limit of normal (ULN), AST (aspartate transaminase) ≤ 2.5 x the ULN
- Serum direct bilirubin ≤ 1.5 ULN) (except in subjects with congenital bilirubinemia, such as Gilbert syndrome, direct bilirubinemia ≤ 1.5 ULN)
- Absolute neutrophil count (ANC) ≥ 1.0 × 109/L
- Platelet count ≥ 75× 109/L (≥ 50× 109/L if myeloma involvement in the bone marrow is > 50%) and no platelet infusion in the 1 week prior to screening platelet count
- Creatinine clearance (CrCl) ≥ 30 mL/minute. Creatinine clearance should be calculated using eGFR (Modified Diet in Renal Disese [MDRD])
- Corrected serum calcium ≤ 13.5 mg/dL (3.4 mmol/L)
- LVEF ≥ 40%. 2-D transthoracic echocardiogram (ECHO) is the preferred method of evaluation. Multigated Acquisition Scan (MUGA) is acceptable if ECHO is not available.
- Females of childbearing potential (FCBP)* complies with the conditions of the Pregnancy Prevention Plan, including confirmation that she has an adequate level of understanding and must agree to ongoing pregnancy testing and to practice contraception or true abstinence. FCBP must use a highly effective and an additional barrier contraception method simultaneously for 4 weeks before starting therapy, during treatment and dose interruptions and for 5 months after the last dose of study drugs.
- Male subjects must agree to practice contraception if sexually active with FCBP during the treatment and for 5 months after the last dose of study drugs. Males must agree to refrain from donating sperm for at least 90 days after the last dose of carfilzomib and for at least 5 months after the last dose of isatuximab.
*Note 1: a FCBP is a woman who:
1) has achieved menarche at some time point,
2) has not undergone a hysterectomy or bilateral oophorectomy or,
3) has not been naturally postmenopausal (amenorrhea following cancer therapy does not rule out childbearing potential) for at least 24 consecutive months (ie, has had menses at any time in the preceding 24 consecutive months).
Note 2: true abstinence is acceptable when this is in line with the preferred and usual lifestyle of the patient. Periodic abstinence (eg, calendar, ovulation, symptothermal, post-ovulation methods) and withdrawal are not acceptable methods of contraception.
Exclusion criteria
- Previous treatment with anti-myeloma therapy (does not include radiotherapy, biphosphonates, or a single short course of steroid ≤ to the equivalent of dexamethasone 40 mg/day for 4 days).
- Patients with non-secretory MM unless serum free light chains are present and the ratio is abnormal or a plasmacytoma with minimum largest diameters of > 2 cm.
- Patients with plasma cell leukemia, amyloidosis, Waldenstrom Disease, POEMS syndrome
- Meningeal involvement of multiple myeloma
- Patient ineligible for autologous transplantation
- Pregnant or lactating females
- Acute active infection requiring treatment (systemic antibiotics, antivirals, or antifungals) within 14 days prior to randomization
- Known human immunodeficiency virus infection (HIV)
- Active hepatitis A, B or C infection. Hepatitis C infection (subjects with hepatitis C that achieve a sustained virologic response after antiviral therapy are allowed), or hepatitis B infection (subjects with hepatitis B surface antigen or core antibody that achieve sustained virologic response with antiviral therapy are allowed). Tests to be performed if required per local country regulations. In fact it is not possible to avoid the risk of virological reactivation with the study treatments.
- Unstable angina or myocardial infarction within 4 months prior to randomization, NYHA Class III or IV heart failure, uncontrolled angina, uncontrolled hypertension, (Uncontrolled hypertension, defined as an average systolic blood pressure ≥ 160 mmHg or diastolic ≥ 100 mmHg despite optimal treatment (measured following European Society of Hypertension/European Society of Cardiology 2013 guidelines), pulmonary embolia, history of severe coronary artery disease, severe uncontrolled ventricular arrhythmias, sick sinus syndrome, or electrocardiographic evidence of acute ischemia or Grade 3 conduction system abnormalities unless subject has a pacemaker
- Non-hematologic malignancy within the past 3 years with the exception of a) adequately treated basal cell carcinoma, squamous cell skin cancer, or thyroid cancer; b) carcinoma in situ of the cervix or breast; c) prostate cancer of Gleason Grade 6 or less with stable prostate-specific antigen levels; or d) cancer considered cured by surgical resection or unlikely to impact survival during the duration of the study, such as localized transitional cell carcinoma of the bladder or benign tumors of the adrenal or pancreas
- Significant neuropathy (Grades 3–4, or Grade 2 with pain) within 14 days prior to randomization as defined by National Cancer Institute Common Toxicity Criteria (NCI CTCAE) 5.0
- Known history of allergy to Captisol® (a cyclodextrin derivative used to solubilize carfilzomib) and to PS80; prior hypersensitivity to sucrose, histidine (as base and hydrochloride salt), or any of the components (active substance or excipients) of study treatments that are not amenable to premedication with steroids, or H2 blockers, that would prohibit further treatment with these agents.
- Contraindication to any of the required concomitant drugs or supportive treatments, including hypersensitivity to all anticoagulation and antiplatelet options, antiviral drugs, or intolerance to hydration due to preexisting pulmonary or cardiac impairment
- Any other clinically significant medical disease or condition that, in the Investigator’s opinion, may interfere with protocol adherence or a subject’s ability to give informed consent
- Pregnant or breastfeeding woman or woman who intends to become pregnant during the participation in the study. FCBP unwilling to prevent pregnancy by the use of 2 reliable methods of contraception for ≥4 weeks before the start of study treatment, during treatment (including dose interruptions), and for at least 28 days following discontinuation of study lenalidomide, or 30 days following discontinuation of carfilzomib or for 5 months after discontinuation of isatuximab treatment, whichever occurs last,
- Male participants who disagree to practice true abstinence or disagree to use a condom during sexual contact with a pregnant woman or a FCBP while participating in the study, during dose interruptions, and for at least 28 days following discontinuation of study lenalidomide, or 30 days following discontinuation of carfilzomib, or for 5 months after discontinuation of isatuximab treatment, whichever occurs last, even if he has undergone a successful vasectomy.